
FDA 510(k) Submission Consulting for Active Medical Devices
Accelerate Your FDA Clearance with Expert 510(k) Consulting for Active Devices
If you’re developing a powered, software-driven, or electro-medical device and targeting the U.S. market, you need to prepare a compliant and accurate 510(k) submission. The FDA 510k process ensures that your device is safe, effective, and substantially equivalent to a legally marketed device. Getting it wrong can mean costly delays.
At Maven Profcon, our specialized FDA 510k consultants help streamline your path to FDA clearance. Whether you’re a startup, global manufacturer, or design firm, our expert team of FDA consultants for medical devices ensures every requirement is met—on time and without compromise.
What Is an FDA 510(k) Submission?
A 510(k) is a premarket submission required by the U.S. FDA for many Class II medical devices, including most active devices like infusion pumps, ECG monitors, or powered surgical tools. To succeed, you must demonstrate substantial equivalence to an existing (predicate) device.
Services by Our FDA Medical Device Consultants
As experienced FDA 510k consultants, we provide end-to-end support, including:
1. Predicate Device Analysis & Laying down a regulatory strategy
- In-depth research to identify suitable predicates and validating the equivalent option.
- Develop a robust strategy tailored to your product type and risk level
2. 510(k) File Preparation
- Technical file aligned with FDA’s current eSTAR or traditional formats
- Labeling, risk analysis, software documentation, and more
3. Performance & Bench Testing Support
- Guidance on protocols, standards (e.g., IEC 60601 series, EMC, Biocompatibility), and FDA expectations
4. Software Validation Support
- Help with software documentation per FDA guidance for SaMD or embedded software
5. Submission & FDA Communication
- We assist with eSTAR or traditional submission preparation
- We act as your dedicated FDA consultant medical device liaison/US Agent
- We handle Interaction with FDA reviewers, RTA responses, deficiencies, and clarification rounds
6. Post-Submission Monitoring
- Ongoing tracking and response planning during the review cycle
Our consultants ensure every section of your submission meets expectations. As FDA medical device consultants, we’ve handled diverse powered and software-integrated products.
Types of FDA 510(k) Submissions for Medical Devices
(And Key Regulatory Requirements Under 21 CFR Part 807)
Understanding the types of FDA 510(k) submissions and compliance obligations like 21 CFR 807 is critical for any medical device manufacturer seeking FDA clearance. Whether you’re a startup or an established manufacturer, choosing the right 510k submission type ensures smoother approval and faster market access. Here’s a breakdown of the three main types of 510(k) submissions most commonly used in the US medical device regulatory process.
1. Traditional 510(k)
The Traditional 510(k) is the most commonly submitted application for FDA clearance of a medical device. It requires detailed documentation to demonstrate substantial equivalence to a legally marketed predicate device.
Best for :
• New medical devices
• Significant design or performance changes
• First-time FDA submissions
As experienced FDA 510k consultants, we guide you through every section of the Traditional 510k — from device description to performance testing and labeling.
2. Special 510(k)
The Special 510(k) is a streamlined route available when you’re making modifications to your own legally marketed device without changing its intended use or core technology.
Best for :
• Design upgrades
• Software updates
• Label changes
• Internal improvements
With our help as experienced FDA medical device consultants, your Special 510k can often be cleared in just 30 days.
3. Abbreviated 510(k)
The Abbreviated 510(k) submission uses FDA-recognized consensus standards or guidance documents to support substantial equivalence — often resulting in a faster review.
Best for :
• Devices covered under FDA-recognized standards
• Firms using validated performance test protocols
Our team of FDA 510k consultants will help you prepare a standards-driven submission that meets all regulatory expectations efficiently.
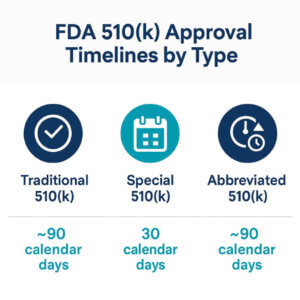
FDA 510(k) Approval Timelines by Type
1. Traditional 510(k)
• Standard FDA Review Time: ~90 calendar days
• Real-World Average: 100–150 days, considering RTA (Refuse to Accept) and AI (Additional Information) cycles.
• Includes full documentation review, often used for new or significantly modified devices.
2. Special 510(k)
• FDA Review Target: 30 calendar days
• Fast-track option for manufacturers modifying their own legally marketed device where intended use and core tech stay the same.
3. Abbreviated 510(k)
• Target Review Time: Similar to Traditional 510(k) (90 days), but may result in slightly faster clearance if standards/guidance are well-aligned.
• Efficiency depends on how well your submission aligns with FDA-recognized standards and guidance documents.
Other Factors That Can Affect Timelines
• FDA’s RTA screening process (initial review within ~15 days)
• AI Letters (requests for Additional Information)
• Quality of your predicate comparison
• Whether device testing is complete and well-documented
• Internal FDA workload and reviewer assignment
Hence it is imperative that expert USFDA 510(k) consultants be at your disposal to significantly reduce these timelines.
- International medical device manufacturers looking for a reliable FDA consultant for medical devices
- Medical device Startups and OEMs developing powered diagnostic or therapeutic devices
- Established or global medical device manufacturers seeking faster, more compliant submissions
- Contract medical device manufacturers supporting U.S. market entry for their clients
Whether you’re preparing your first or fifth 510(k), our FDA consultants for medical devices help you get there faster—with less risk and better results.
- Experienced 510(k) Consultants – Years of experience with a range of powered/active medical devices
- U.S.-Focused Expertise – We speak the language of the FDA with a proven track record as a US FDA consultant for global firms.
- Faster Time to Clearance – Avoid delays or RTA rejections
- Complete Support – Complete technical, testing, and submission support
1. Free Consultation & Device Assessment – Assess your product and determine classification
2. Regulatory Strategy & Predicate Search – With insight from expert FDA medical device consultants
3. Documentation & Testing – IEC 60601, EMC, software, and performance reports
4. Drafting & Internal QA – Full submission file prepared by our 510k consultants
5. Submission & FDA Response – Support through FDA communication and reviews
6. Final Clearance – You receive your FDA 510(k) clearance and go to market
Get FDA Clearance with Confidence
Don’t let documentation errors or misclassification delay your product launch. Let Maven Profcon be your trusted partner. We’re a dedicated team of FDA 510k consultants, ready to support your journey with precision and professionalism.
Frequently Asked Questions
Typically 12-15 months, depending on device complexity and FDA review timelines. With Maven Profcon’s FDA 510k consultants, we help reduce delays by ensuring accuracy up front.
Yes – Our FDA medical device consultants provide guidance on applicable standards and test planning.
Absolutely. As US FDA consultants, we routinely work with global manufacturers preparing for U.S. market entry.